(информация для педагогов)
Причины
Синдром Прадера-Вилли- врожденное заболевание, при котором возникает сочетание ожирения, низкого роста, снижения функции половых желез (гипогонадизм) и низкого интеллекта. Это заболевание имеет очень широкий спектр проявлений и признаков. Течение болезни отличается в каждом отдельном случае и может варьировать от легкой формы до тяжелой, которая прогрессирует в течение всей жизни человека.
Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г. и встречается у 1 человека на 10000-25000 новорожденных (мальчиков и девочек одинаково). Причиной данного генетического заболевания является отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15 отцовской хромосоме. Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы.
Клиника
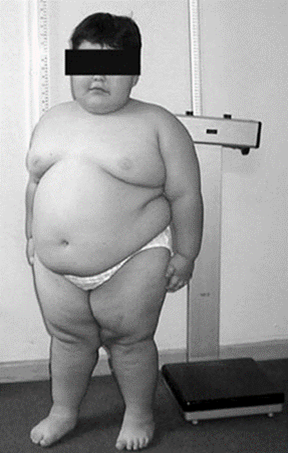
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание. Заболевание характеризуется выраженной мышечной гипотонией при рождении, сохраняющейся в течение первого года жизни ребенка. Сосательный и глотательный рефлексы снижены, что затрудняет кормление ребенка. Из-за гипотонии у таких детей задерживается развитие двигательных функций: они с трудом учатся держать голову, сидеть и т. д. Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает.
Позднее, ко второму-четвертому году жизни появляются постоянное чувство голода и отсутствие насыщения, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в верхних отделах конечностей. Из-за тяжелого ожирения грозным осложнением является остановка дыхания во сне.
Рост больных нередко снижен. Часто отмечается удлиненная форма головы, миндалевидный разрез глаз, низко расположенные ушные раковины, широкая переносица, маленький рот с тонкой верхней губой. Стопы и кисти больных диспропорционально маленькие. У 75% детей наблюдается слабая пигментация кожи, волос и радужки.
У мальчиков и девочек при рождении отмечается недоразвитие половых органов. В дальнейшем заболевание проявляется задержкой или отсутствием полового созревания, бесплодием.
Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед.). Как правило, дети с синдромом Прадера-Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарем, но их собственная речь обычно хуже, чем понимание. Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже.
У большинства людей с синдромом Прадера — Вилли наблюдается задержка психического и речевого развития. Согласно исследованиям, которые провели Л. М. Керфс и Дж. П. Фринс (1992)[5],
- 5 % обследованных продемонстрировали средний уровень коэффициента интеллекта (более 85 баллов по шкале IQ);
- 27 % — уровень на грани среднего (70-85 баллов);
- 34 % — уровень слабого отставания (50-70 баллов);
- 27 % — уровень среднего отставания (35-50 баллов);
- 5 % — сильное отставание (20-35 баллов);
- менее 1 % — значительное отставание.
Дети с таким синдромом доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, косоглазие. Основные психические расстройства, возникающие у больных СПВ - это подергивания за кожу и повышенная тревожность. Психиатрические симптомы, например, такие как галлюцинации, паранойя и депрессия, которые могут возникать примерно в 5-10% больных молодых людей. Психиатрические и поведенческие проблемы являются наиболее частой причиной госпитализации больных с СПВ.
Продолжительность жизни больных может достигать 60 лет и более. Нередко у таких детей развивается сахарный диабет.
Лечение
Синдром Прадера-Вилли является врожденной генетической аномалией и, следовательно, не может быть излечен. Однако если диагностировать данное заболевание на раннем этапе и начать его лечение, то прогноз развития заболевания становится более оптимистичным.
Младенцы со сниженным мышечным тонусом должны получать массаж и другие виды специальной терапии. Комплекс лечебных мероприятий включает также диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины). Рекомендуется терапия гормоном роста.
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом
Медико-генетическое консультирование
Родителям ребенка с синдромом Прадера-Вилли рекомендуется пройти генетическое обследование, прежде чем планировать дальнейшую беременность, поскольку существует риск того, что следующий ребенок у тех же родителей родится также с синдромом Прадера-Вилли, что зависит от механизма, вызвавшего генетический сбой.
Общество и культура
Впервые публичная информация о синдроме Прадера-Вилли появилась в британских СМИ в июле 2007 года, когда телевизионный канал Channel 4 показал программу под названием Can't Stop Eating («Не могу прекратить есть»), в которой описывалась ежедневная жизнь двух человек с синдромом Прадере-Вилли.
С помощью родителей, школы, врачей и друзей люди с СПВ могут выполнять те же действия, что и их здоровые сверстники . Они могут учиться, заниматься своими хобби, найти работу и стать независимыми.
Источники